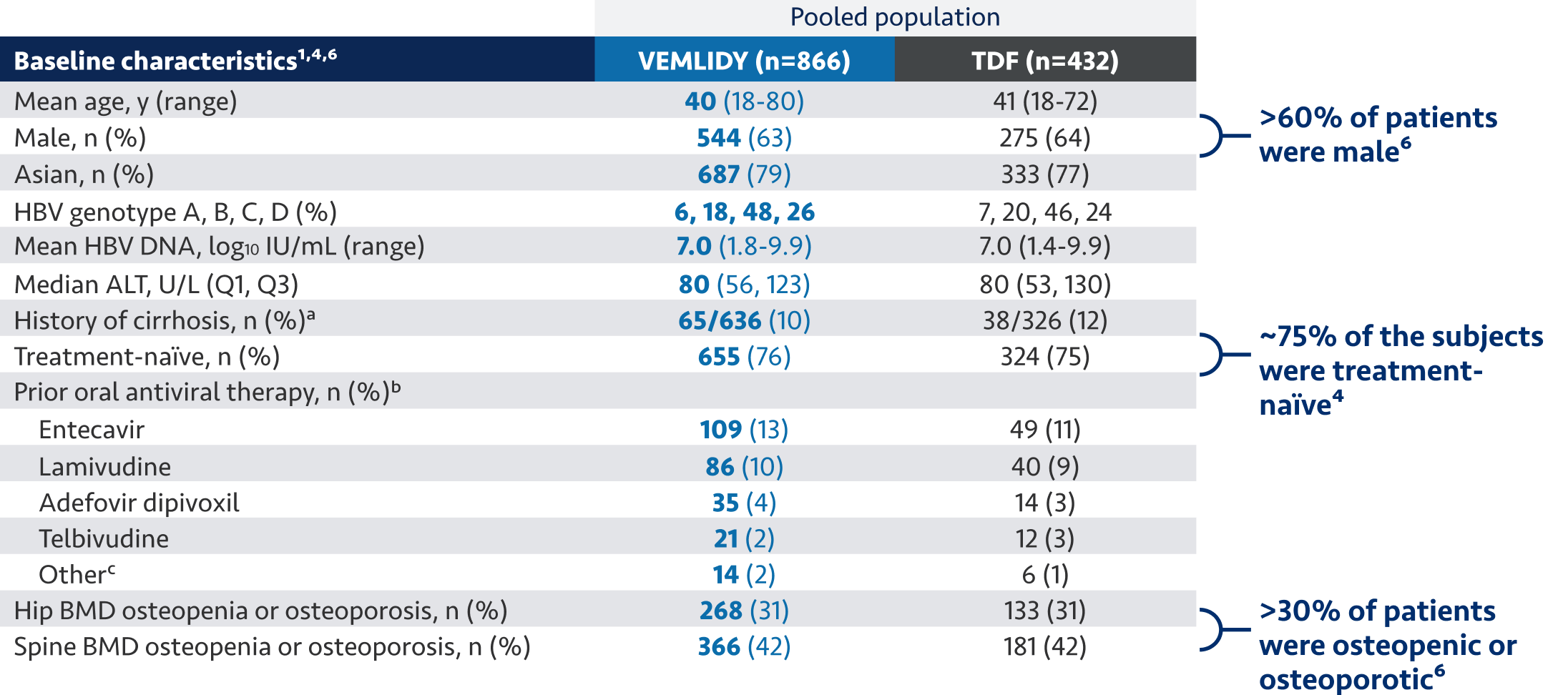
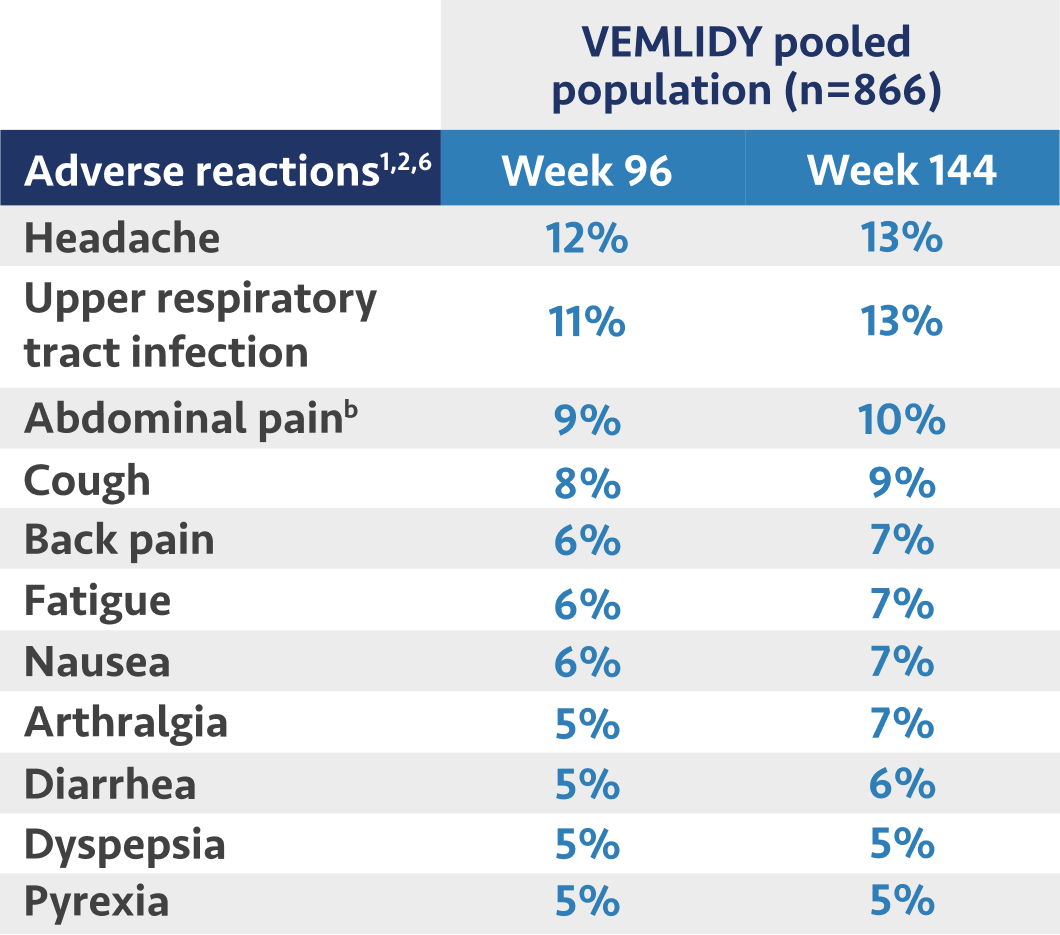
VEMLIDY demonstrated a well-established safety profile for a broad range of patient types through 8 years1-4
Trials 108 and 110 (pooled)
Adverse reactionsa (all grades) reported in ≥5% of patients on VEMLIDY in Trials 108 and 110 (Week 96 and Week 144 analyses)
Differences were observed between VEMLIDY and TDF in certain lipid parameters1-3
In Trials 108 and 110:
- Week 96: Mean changes from baseline in low-density lipoprotein cholesterol (LDL-C; fasted) and triglycerides (TG; fasted) were +7 mg/dL and +13 mg/dL for VEMLIDY, versus -10 mg/dL and -7 mg/dL for TDF1
- Week 144: Mean changes from baseline in LDL-C (fasted) and TG (fasted) were +8 mg/dL and +18 mg/dL for VEMLIDY, versus -8 mg/dL and -2 mg/dL for TDF2
- Week 384: Among patients receiving VEMLIDY, the median change in LDL-C (fasted) was +16 mg/dL and TG (fasted) was +9 mg/dL. Among patients who switched from TDF to VEMLIDY at Week 96, the median change in LDL-C (fasted) was +17 mg/dL and TG (fasted) was +11 mg/dL. Among patients who switched from TDF to VEMLIDY at Week 144, the median change in LDL-C (fasted) was +11 mg/dL and TG (fasted) was +14 mg/dL3
In Trial 4018:
- Week 48: Changes from baseline in lipid parameters in the VEMLIDY and TDF groups were similar to those observed in Trials 108 and 110 at Week 961
- Week 96: Lipid parameters in patients who were switched from TDF to VEMLIDY at Week 48 were similar to those of patients who continued on VEMLIDY2
8-year safety data
Pooled Safety Analysis (Week 384): Pooled safety analysis (observed data) from Trials 108 and 110 was assessed at Week 384. This analysis includes 866 patients who initiated VEMLIDY at baseline, 207 patients who switched from TDF to VEMLIDY at Week 96, and 225 patients who switched from TDF to VEMLIDY at Week 144.3
Most common ARs (all grades) reported in ≥5% of patients on VEMLIDY who entered the open-label extension (Week 384 analysis)
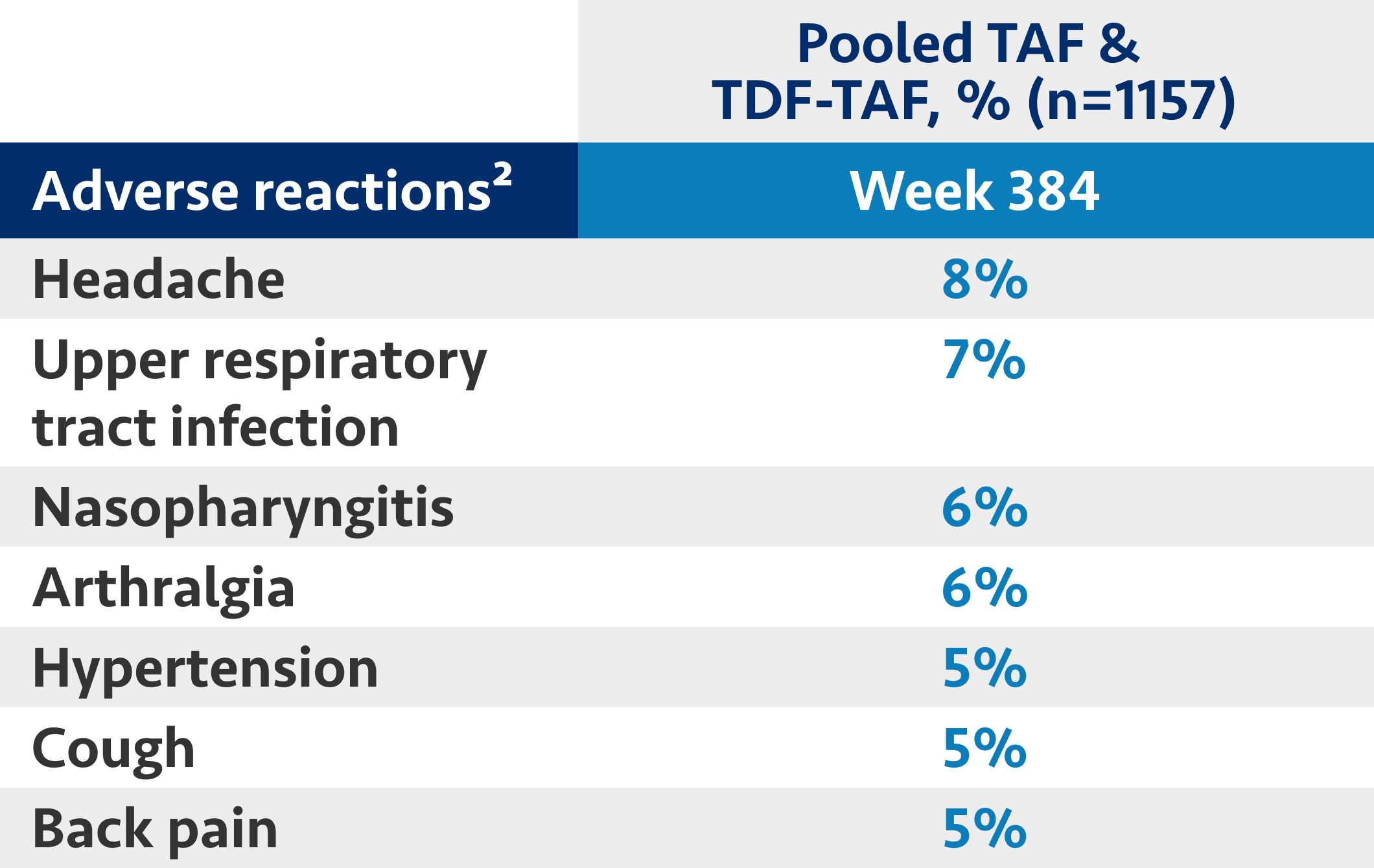
The 8-year analysis is not presented in the VEMLIDY full Prescribing Information.
HCC surveillance in Trials 108 and 110 through Week 384
At Week 384, there were a total of 21 cases (1.6% incidence) of hepatocellular carcinoma (HCC) observed in Trials 108 and 110.3,a,c

HCC surveillance was included as part of the 96-week protocol amendments for Trials 108/110. These trials were not powered to look at any treatment effect on HCC and no results should be drawn based on these observations. This information is not in the VEMLIDY Prescribing Information.2
aFrequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug.1
bGrouped term including abdominal pain upper, abdominal pain, abdominal pain lower, and abdominal tenderness.1
c3 cases of HCC were observed in the open-label TDF➔VEMLIDY group, all of which developed before Week 48 of the open-label phase.2
Use of VEMLIDY in special populations1
Pregnancy
- There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VEMLIDY during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263
- Available data from the APR show no statistically significant difference in the overall risk of birth defects for tenofovir alafenamide (TAF) compared with the background rate for major birth defects of 2.7% in the US reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP). The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the US general population is 15% to 20%
- Based on prospective reports to the APR of over 1330 exposures to TAF-containing regimens during pregnancy resulting in live births (including over 1080 exposed in the first trimester and over 240 exposed in the second/third trimester), the prevalence of birth defects in live births was 3.9% (95% CI: 2.8% to 5.2%) and 4.8% (95% CI: 2.5% to 8.3%) following first and second/third trimester exposure, respectively, to TAF-containing regimens. Methodological limitations of the APR include the use of MACDP as the external comparator group. The MACDP population is not disease-specific, evaluates women and infants from a limited geographic area, and does not include outcomes for births that occurred at less than 20 weeks’ gestation
Lactation
- Data from the published literature report the presence of TAF and tenofovir in human milk. Data from the published literature have not reported adverse effects of TAF on a breastfed child. There are no data on the effects of TAF on milk production
- The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for VEMLIDY and any potential adverse effects on the breastfed infant from VEMLIDY or from the underlying maternal condition
Geriatric use
- VEMLIDY has been administered to 89 patients aged 65 and over in clinical trials, and no differences in safety or efficacy have been observed between these patients and younger patients
Patients with ESRD
- No dosage adjustment is needed for patients with ESRD (eCrCl <15 mL/min) receiving chronic hemodialysis
- In patients on chronic hemodialysis, on hemodialysis days, administer VEMLIDY after completion of hemodialysis treatment
- VEMLIDY is not recommended in patients with ESRD who are not receiving chronic hemodialysis
IMPORTANT SAFETY INFORMATION
BOXED WARNING: POSTTREATMENT SEVERE ACUTE EXACERBATION OF HEPATITIS B
- Discontinuation of anti-hepatitis B therapy, including VEMLIDY, may result in severe acute exacerbations of hepatitis B. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy, including VEMLIDY. If appropriate, resumption of anti-hepatitis B therapy may be warranted.
Warnings and Precautions
- Risk of Development of HIV-1 Resistance in HBV/HIV-1 Coinfected Patients: Due to this risk, VEMLIDY alone should not be used for the treatment of HIV-1 infection. Safety and efficacy of VEMLIDY have not been established in HBV/HIV-1 coinfected patients. HIV antibody testing should be offered to all HBV-infected patients before initiating therapy with VEMLIDY, and, if positive, an appropriate antiretroviral combination regimen that is recommended for HBV/HIV-1 coinfected patients should be used.
- New Onset or Worsening Renal Impairment: Postmarketing cases of renal impairment, including acute renal failure, proximal renal tubulopathy (PRT), and Fanconi syndrome have been reported with TAF-containing products. Patients with impaired renal function and/or taking nephrotoxic agents (including NSAIDs) are at increased risk of renal-related adverse reactions. Discontinue VEMLIDY in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome. Monitor renal function in all patients – See Dosage and Administration.
- Lactic Acidosis and Severe Hepatomegaly with Steatosis: Fatal cases have been reported with the use of nucleoside analogs, including tenofovir disoproxil fumarate (TDF). Discontinue VEMLIDY if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity develop, including hepatomegaly and steatosis in the absence of marked transaminase elevations.
Adverse Reactions
Most common adverse reactions (incidence ≥5%; all grades) in clinical studies through week 144 were headache, upper respiratory tract infection, abdominal pain, cough, back pain, arthralgia, fatigue, nausea, diarrhea, dyspepsia, and pyrexia.
Drug Interactions
- Coadministration of VEMLIDY with drugs that reduce renal function or compete for active tubular secretion may increase concentrations of tenofovir and the risk of adverse reactions.
- Coadministration of VEMLIDY is not recommended with the following: oxcarbazepine, phenobarbital, phenytoin, rifabutin, rifampin, rifapentine, or St. John’s wort. Such coadministration is expected to decrease the concentration of tenofovir alafenamide, reducing the therapeutic effect of VEMLIDY. Drugs that strongly affect P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) activity may lead to changes in VEMLIDY absorption.
Consult the full prescribing information for VEMLIDY for more information on potentially significant drug interactions, including clinical comments.
Dosage and Administration
- Testing Prior to Initiation: HIV infection.
- Prior to or When Initiating, and During Treatment: On a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus.
- Dosage in Adults: 1 tablet taken once daily with food.
- Renal Impairment: Not recommended in patients with end stage renal disease (ESRD; eCrCl <15 mL/min) who are not receiving chronic hemodialysis; in patients on chronic hemodialysis, on hemodialysis days, administer VEMLIDY after completion of hemodialysis treatment.
- Hepatic Impairment: Not recommended in patients with decompensated (Child-Pugh B or C) hepatic impairment.
Pregnancy and Lactation
- Pregnancy: A pregnancy registry has been established for VEMLIDY. Available clinical trial data show no significant difference in the overall risk of birth defects for VEMLIDY compared with the background rate of major birth defects in the U.S. reference population.
- Lactation: TAF and tenofovir can pass into breast milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for VEMLIDY and any potential adverse effects on the breastfed infant from VEMLIDY or from the underlying maternal condition.
INDICATION
VEMLIDY is indicated for the treatment of chronic hepatitis B virus (HBV) infection in adults with compensated liver disease.
Please see full Prescribing Information for VEMLIDY, including BOXED WARNING.